
Prions
What they are:
Infectious proteins
In the 1950s and ‘60s, an epidemic of a disease known as kuru hit the Fore tribal peoples in Papua New Guinea. Victims experienced muscle tremors that eventually gave way to a complete loss of motor control. The late stages of the illness brought on uncontrolled emotional outbursts, leading some to nickname kuru “the laughing death.” Without exception, the disease was fatal. Researchers looking to contain the outbreak realized kuru was being spread through the practice of cannibalism. The Fore people, as part of their funeral rites, ceremonially ate their dead.
Avoiding kuru seems simple, and indeed, when the Fore people abandoned cannibalism, the spread of disease stopped. But in the 1980s and ‘90s a similar scare occurred in England. Mad cow disease affected cattle the same way Kuru affected humans: infected cattle would lose control of their muscles and eventually die. Of course, cows aren’t typically cannibals, but the beef industry used bone meal in cattle feed, meaning the disease could spread in the same way kuru did. But the disease didn’t only affect cows: humans who ate infected meat developed a kuru-like disease. And perhaps most scary: unlike virtually any other disease, the meat remained infectious even after it was cooked.
It turns out that kuru and mad cow disease are not contagious in the way other known diseases are contagious. Every other type of known infectious illness is caused by a virus, bacterium, fungus, or parasite. These are agents that spread by replicating themselves using the instructions encoded in a genome made of DNA or RNA, and they can be killed or inactivated with heat. But transmissible spongiform encephalopathies” (TSEs) like kuru and mad cow disease are unlike other infectious diseases. They pass without any microorganism or genetic information. They are caused by nothing more than an organism’s own improperly folded proteins – also known as prions.
How they cause disease:
Contagious misfolding
Proteins are strings of amino acids that twist and fold into very specific three-dimensional forms. This three-dimensional shape is what allows the protein to do its job. For example, a protein that serves as an enzyme needs to have a very specific shape in order to bind only to its substrate and not to lookalike molecules. When a protein is misfolded, its shape is disrupted and it is unable to do its job. Almost all proteins have the potential to misfold or break down over time, and cleaning them up is part of a cell’s normal housekeeping routine.
So, misfolding makes a prion nonfunctional. But what makes it infectious? Unlike ordinary proteins that happen to misfold, prions can trigger misfolding in the normal versions of that same protein. The result is a chain reaction: just one misfolded prion can cause many of its normally folded protein counterparts to misfold, and each of those can in turn convert many other normally folded prion proteins. Through this process, misfolding spreads exponentially. It’s not unlike the spread of disease in a zombie movie: by recruiting just a few victims who then go on to hunt their own victims, a single zombie can transform an entire town into an undead army in no time.
Most of the known prion diseases are caused by misfolding of a specific protein that scientists named “prion protein” or PrP. Virtually all mammals carry a PrP gene, which is why prion diseases affect all sorts of different animals. Mad cow-disease in cattle, scrapie in sheep, chronic wasting disease in deer and Creutzfeldt-Jakob disease or kuru in humans: these are all essentially the same disease caused by misfolding of PrP. This also explains why some versions of the disease can jump from one species to another, as when humans become infected with mad cow disease after eating tainted beef.
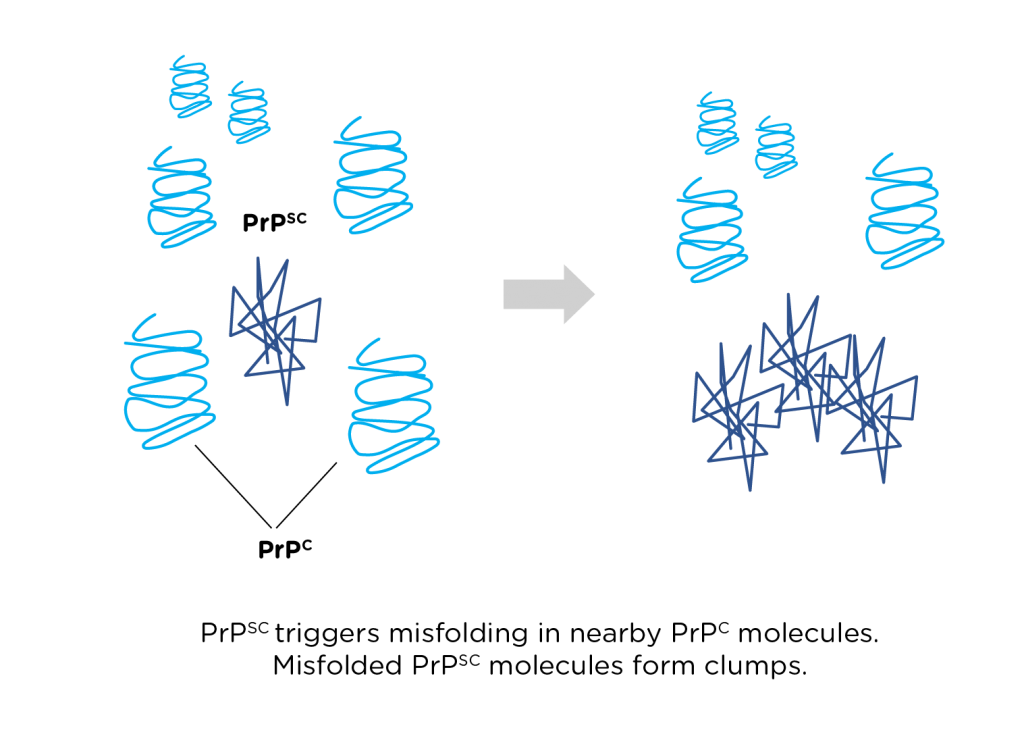
The function of PrP is largely unknown, but we know that in a healthy organism, normally folded PrP can be found along the outside of nerve cell membranes. When normal PrP (sometimes called PrPc) gets converted to misfolded PrP (or PrPsc – the “sc” stands for scrapie), the misfolded molecules clump together. The growing clusters of PrPsc crowd out and eventually kill nearby neurons, leading to the problems with motor control and emotion that we see in patients with TSEs. Because there is currently no way to stop or reverse the spread of misfolding, TSEs are invariably fatal.
Detection & prevention:
You can’t kill what isn’t alive
Testing for a disease caused by a misfolded protein is tough. With other infectious diseases, a doctor or scientist can culture infected samples in a Petri dish to see if colonies of infectious microbes grow, or search for DNA sequences characteristic of disease-causing pathogens. But because prions aren’t alive, they can’t be grown in culture like a disease-causing microbe. And because they don’t carry DNA or RNA, you can’t use genetic tests to look for them. Scientists have had to develop brand-new tests to detect prions. They typically involve incubating normally folded PrPc with a test sample of potentially diseased tissue, then seeing if clumps of PrPsc form.
Even if we’ve developed ways to detect these unusual infectious molecules, preventing their spread is another story. Our tried-and-true ways of preventing disease are things that kill infectious microbes: we use disinfectants and cook our food. These methods don’t work on prions; they aren’t alive, so they can’t be killed with our normal disinfectants or heat. This is a particularly sobering fact given that prion diseases are incurable.
Currently, our only way of preventing prion disease is avoiding contact with infected tissue. New feeding practices and vigilant monitoring have meant there has not been an outbreak of mad cow disease in many years, and some U.S. communities have opened testing centers where deer hunters can have their prey tested for chronic wasting disease before they eat it. But these measures alone are no solution; we are still unable to cure those who come down with a prion disease. The scientific community is crying out for new research on prions. Only by learning more about these dangerous molecules can we hope to engineer weapons against them.
Questions
Review:
- Some disinfectants work by rupturing cell membranes of bacteria and other harmful microorganisms. Why won’t these disinfectants work on prions?
- Plenty of proteins will randomly misfold and become nonfunctional, but most misfolded proteins don’t become prions. What makes a prion more dangerous than just any misfolded protein?
- Why is it fair to say kuru, mad cow disease, scrapie, and chronic wasting disease are all the same disease, even though they go by different names?
- How does the clumping up of misfolded PrP molecules lead to the neurological symptoms (loss of muscle control, emotional outbursts) observed in victims of prion diseases?
- With most infectious illnesses, doctors can use genetic tests to determine what pathogen is making someone sick. Why can’t a doctor use genetic tests to test for a prion disease?
Critical thinking:
- Sickle cell anemia is a disease that is caused by a mutation in a person’s beta-hemoglobin gene. When the mutation is present, the hemoglobin proteins in the cell will clump together. This clumping of proteins is what leads to typical sickle cell disease symptoms. How is sickle cell similar to prion disease? How is sickle cell disease different?
- Creutzfeldt-Jakob disease is a rare disease caused by PrP misfolding. It kills about 350 people a year in the United States. Of course, these people are not cannibals and have not contracted the disease through contact with infected tissues. How can this be? Propose how someone might come down with a prion disease without having caught it from another person.
Discussion:
- Medical professionals often use the word “colonized” when referring to a person or animal who has contracted an infectious disease. Would it be right to say a person with a TSE has been colonized by the disease-causing agent? Explain your answer.
Answer key:
Available to teachers upon request: dnadots@minipcr.com