

RNA-Seq (RNA sequencing)
What it is:
Using sequencing to measure RNA
Seeing an artisan’s toolkit can tell you a lot about how that person works. But to truly understand their process, you would want to see how and when all those different tools are used. In genetics, you can think of an organism’s genome as its genetic toolkit. Sequencing that genome will give you a lot of information about the tools available to the organism, but it won’t give you a full picture of under what circumstances each of those different tools is used. Studying how and when organisms use their genetic toolkit is key to understanding how various tissues and cells function differently, how diseased cells behave differently from normal cells, and how organisms respond to their environment at the molecular level. To understand these things, we must know under what conditions each individual gene is transcribed into RNA. For a long time now, scientists have approached this problem by measuring single specific messenger RNAs using reverse-transcription qPCR, while microarrays have given scientists the ability to quantify thousands of RNA transcripts simultaneously. But both these techniques require researchers to know in advance the sequences of RNA that they are trying to quantify, and they are limited in the amount of sequences that can be accurately investigated at once. In other words, you have to know what you are looking for before you can find it, and even then, the picture they offer is limited.
But now, the same new technologies that are allowing researchers to sequence whole genomes faster and more affordably than ever are being turned to RNA in a process known as RNA-Seq (pronounced RNA–seek). In doing so, scientists can record the nucleotide sequence, the order of the As, Us, Gs, and Cs, of nearly every RNA molecule in a cell at one particular time. This provides a snapshot of all the cellular instructions being sent out at a particular time. And because all the RNA in a cell can be sequenced without first having to design a specific probe, scientists can even find sequences that they did not know existed. Furthermore, they can observe variation in RNA that would not be possible using older techniques. And by using computational methods to count how many times a particular stretch of RNA is sequenced, a researcher can establish how much that particular gene is used compared to others. In essence, they can see exactly how much every tool in the toolkit is being used at any one time.
How it works:
Next-gen sequencing turned to RNA
So-called next-generation sequencing has revolutionized scientists’ ability to gather and analyze large amounts of sequence data quickly and affordably. Using next-gen sequencers scientists can read hundreds of millions of nucleic acid fragments simultaneously, each sequence up to a couple hundred base pairs in length. These millions of short sequences are then lined up by matching overlapping sections to construct the much longer original sequence. In applying this technology to RNA, there are a few different methods, but most follow the same basic approach.
Researchers first isolate the nucleic acids in a sample and eliminate all of the DNA, usually using an enzyme. Of the RNA that remains, a significant majority, usually well over 90%, is ribosomal RNA (rRNA) which is of little interest for most studies, so researchers first work to make the type of RNA they are interested in relatively more abundant. If researchers are working on eukaryotes and are only interested in messenger RNA (mRNA), they can select for sequences that have a poly-adenine tail, a modification present in all eukaryotic mRNA. If only small RNA, like micro RNA, is desired, scientists can select sequences based on size, and so on. The isolated RNA is then broken down into smaller pieces that the sequencer can process. These smaller fragments of RNA are then copied into DNA in a process known as reverse transcription. The resulting molecule is known as complementary DNA or cDNA. A nucleotide tag is attached to the end of the cDNA fragments, a step necessary for the sequencer to recognize the sample. Then the entire sample, hundreds of millions of cDNA fragments, are sequenced simultaneously.
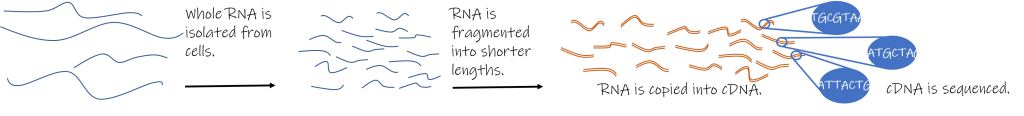
RNA-Seq results will be in the form of a list of hundreds of millions of sequences read by the sequencing machine. A computer will then organize the different short sequences and line them up, piecing together the individual fragments to reconstruct the longer RNA sequences that the process started with. This will provide a nearly complete record of every RNA molecule in a cell at one time. By counting how many times each specific sequence is represented in the data, it will also show how common or rare specific RNA sequences are. Matching these RNA sequences to known genes will tell you how much all the different tools in the toolkit are used by the organism under different conditions.
The future:
Direct, longer, more precise
RNA-Seq has given scientists a tool that can investigate RNA more precisely than ever before. For example, if after transcription the RNA sequence is modified in any way, sections removed or reordered, RNA-seq will be able to identify those differences—something that was difficult with earlier methods. But because the cDNAs are usually broken into smaller pieces before sequencing, recognizing rare or subtle modifications can still be difficult. A new generation of sequencing technology can read much longer sequences, though. Now entire mRNA molecules can be sequenced in a single go, with no need to fragment the samples. This can be incredible powerful when trying to look for rare mRNA sequence variants or less common modifications made to the molecule. And new nanopore sequencing technology can even sequence RNA directly without first transcribing it to cDNA, helping to eliminate possible biases and artifacts that are sometimes introduced in the reverse transcription step.
New methods are also allowing researchers to investigate the expression patterns of single cells instead of whole tissues. So-called scRNA-Seq can give scientists insights into how individual cells within a tissue may behave differently from each other. In 2018, this led to researchers discovering a whole new cell type present in mouse and human respiratory tracts that was previously unknown to science. It turns out that this new cell type plays an important role in cystic fibrosis. The study illustrates the power of RNA-seq to reveal how genetically identical cells behave differently—even when you don’t know exactly what you are looking for.
Questions
Review:
- qPCR is often considered the gold standard for measuring gene expression of a single gene. What are some advantages to RNA-Seq vs qPCR?
- How does sequencing all the RNA in a cell tell you how much a particular gene was used?
- Why do researchers need to enrich their RNA samples for a particular type of RNA, Why not just sequence all of it?
- In what ways would longer sequencing reads be more informative than many short reads?
- Why can sequencing individual cells be more informative than sequencing whole tissues?
Critical thinking:
- Fragmenting a sample for next-gen sequencing can make it difficult to reconstruct rare sequences. Why would it be easier to reconstruct more common sequences, but not rare ones?
- Direct sequencing of RNA (without a reverse transcription step) requires more RNA than a can be obtained by a single cell. Given the stated advantages of both long direct reads and single cell sequencing, under what conditions may you decide to use one approach over the other?
Discussion:
- RNA-Seq allows scientists to study an organism’s RNA transcripts, sometimes called its transcriptome, without having to even know its genome first. Deciphering an organism’s genome, it’s entire DNA sequence, is often held up as the key to understanding its biology. But if you could only know one, which do you think would be more informative, an organism’s genome or its transcriptome? Defend your answer.
Answer key:
Available to teachers upon request: [email protected]